Proposal/Future Directions
As a final part of our project, we were tasked to come up with experiments that would further progress the current understanding of our gene of interest; preferably from the data we have accumulated throughout the semester. I chose to do two different approaches in that regard. My first set of proposed experiments addresses a current gap in the understanding of Sox9 I deduced from reading the literature. The second aim I am proposing is indeed something I have found in my own data that I would have wanted to address. Of course, this is assuming that I had the means to actually perform the experiments... which I do not, unfortunately.
Specific Aim 1: in vivo DNA binding sites of Sox9
For my first proposed set of experiments, the main goal is to determine the cis-regulatory elements (CREs) that Sox9 binds to in vivo during skeletal development, and in particular, chondrogenesis. The modern method to finding punative CREs/transcription factor binding sites is to perform Chromatin Immunoprecipitation coupled with Ultra High-Throughtput Massively Parallel DNA Sequencing, or "ChIP-seq" (Johnson et al 2007; Jothi et al 2008; Please refer to the figure to the right for a summary of ChIP-seq). The previous method used was known as ChIP-chip, which used tiled arrays instead of sequencing, and is not as accurate as ChIp-seq.
It should be noted that there have been a previous study, Oh et al 2010, that looked for Sox9 binding sites using a genomic method of ChIP. It is undeniable that this study was very informative in understanding Sox9's transcriptional activity. However, this study used the more archaic method of ChIP-chip, selected specific candidate probes for genes which instills incredible bias to what one finds, and used a rat chondrosarcoma cell line, which doesn't reflect the true in vivo (and thus physiologically relevant) nature of the developing skeleton. I would therefore propose to test for the in vivo binding of Sox9 in mouse embryonic limb mesenchyme and to perform a completely unbiased approach to find potential Sox9 binding sites throughout chondrogenesis in the developing limb in the hope to
It should be noted that there have been a previous study, Oh et al 2010, that looked for Sox9 binding sites using a genomic method of ChIP. It is undeniable that this study was very informative in understanding Sox9's transcriptional activity. However, this study used the more archaic method of ChIP-chip, selected specific candidate probes for genes which instills incredible bias to what one finds, and used a rat chondrosarcoma cell line, which doesn't reflect the true in vivo (and thus physiologically relevant) nature of the developing skeleton. I would therefore propose to test for the in vivo binding of Sox9 in mouse embryonic limb mesenchyme and to perform a completely unbiased approach to find potential Sox9 binding sites throughout chondrogenesis in the developing limb in the hope to
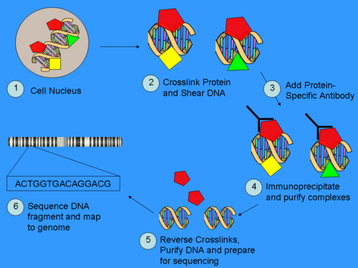
First, living cells/tissue are crosslinked using paraformaldehyde, then followed by the shearing of chromatin into short fragments of about 0.2-1kb in length. The protein-DNA cross-linked fragments are then immunoprecipitated using a protein-specific antibody. Finally, the cross-linked DNA and proteins are reversed, and the DNA is subsequently purified and assayed to determine the sequence bound by that protein. The position and occupancy can also be determined using the data. (Johnson et al 2007; Jothi et al 2008; image taken from http://en.wikipedia.org/wiki/ChIP-Seq)
(1) find Sox9 binding sites during in vivo chondrogenesis, which should confirm many binding sites found in Oh et al 2010 (only with higher resolution), and to potentially find novel genes regulated by Sox9 binding to their representative CREs.
(2) Next, I would want to find what different CREs Sox9 binds to at different embryonic stages, or if Sox9 is homeostatic in what it binds to throughout its normal developmental progression.
(3) I also would like to see what happens to the Sox9 transcriptional binding landscape when the characteristic Campomelic Dysplasia haploinsufficiency of Sox9. Would Sox9 not be able to bind to certain CREs if there is not enough Sox9 to properly dimerize? Or would Sox9 bind to different CREs in the haploinsufficient state, and would these potentially change what happens downstream and ultimately allow the CM phenotype to arise? Would it be simply just Sox9 binding with less affinity to its target CREs? Or is it a combination of all of these?
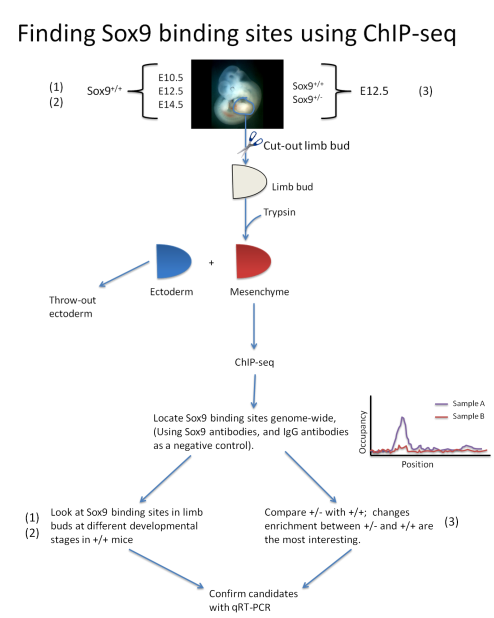
These questions are of great interest to me, and a way to answer these questions in vivo is a difficult one. Therefore, I thought up of an experimental design (diagrammed below) that will hopefully be a good system to find physiologically relevant Sox9 binding sites during chondrogeneis. The experiment relies on the fact that chondrogenesis occurs in the mesenchyme (with ectoderm being irrelevant), so a mild treatment of trypsin to the limb bud removed from the embryo at the described stages and genotypes would cause the ectodermal layer to be dissociated from the mesenchyme. This should minimize uninformative background Sox9 binding. The mesenchyme would then be subject to the ChIP-seq protocol using Sox9 antibodies to determine Sox9 binding sites in the different experimental samples. These samples would then be compared to each other in the way described below to answer questions 1, 2, and 3. Finally, another set of samples duplicated from before will be subject to RT-PCR using probes of the candidate genes found to reconfirm the promoter/enhancer activity of the Sox9 binding sites found.
(2) Next, I would want to find what different CREs Sox9 binds to at different embryonic stages, or if Sox9 is homeostatic in what it binds to throughout its normal developmental progression.
(3) I also would like to see what happens to the Sox9 transcriptional binding landscape when the characteristic Campomelic Dysplasia haploinsufficiency of Sox9. Would Sox9 not be able to bind to certain CREs if there is not enough Sox9 to properly dimerize? Or would Sox9 bind to different CREs in the haploinsufficient state, and would these potentially change what happens downstream and ultimately allow the CM phenotype to arise? Would it be simply just Sox9 binding with less affinity to its target CREs? Or is it a combination of all of these?
These questions are of great interest to me, and a way to answer these questions in vivo is a difficult one. Therefore, I thought up of an experimental design (diagrammed below) that will hopefully be a good system to find physiologically relevant Sox9 binding sites during chondrogeneis. The experiment relies on the fact that chondrogenesis occurs in the mesenchyme (with ectoderm being irrelevant), so a mild treatment of trypsin to the limb bud removed from the embryo at the described stages and genotypes would cause the ectodermal layer to be dissociated from the mesenchyme. This should minimize uninformative background Sox9 binding. The mesenchyme would then be subject to the ChIP-seq protocol using Sox9 antibodies to determine Sox9 binding sites in the different experimental samples. These samples would then be compared to each other in the way described below to answer questions 1, 2, and 3. Finally, another set of samples duplicated from before will be subject to RT-PCR using probes of the candidate genes found to reconfirm the promoter/enhancer activity of the Sox9 binding sites found.
Since Oh et al 2010 indeed found Sox9 binding sites, one would expect to find some of the candidate binding sites to be found again, more or less. I predict that most of the discovered binding sites in that study would appear again. For example, Col2a1, syndecan-3, Sox5, Osx, Runx2, Aggrecan, Patched, BMP4, etc. It would also be interesting to see if Sox9 is found to be bound to in my proposed experimental design that were not found in Oh et al 2010. Such candidates would include Beta-Catenin, Sox6, PGC-alpha, Wnt9, etc. Of course, the most interesting genes I would like to see would be the novel genes that were either not previously known to be involved in chondrogenesis or have absolutely nothing known about its molecular/biological function.
Specific Aim 2: Role of potential Nanog - Ctnnb1 (Beta-Catenin) - Sox9 interaction
Upon using the MEME motif finding algorithm, I found many interesting motifs. However, the most interesting motif I found was a Nanog-binding motif found at the 3' end of the Sox9 gene locus. This motif is found in both mice and humans, and has a high-likelihood of existing. Therefore, I decided that I should look into this a bit further. I found that in addition to Nanog's well known-role in maintaining pluripotency (Mitsui et al 2003), it has also been shown to maintain chondrocyte differentiation in vitro (Zheng et al 2010). However, the mechanism is not understood on how this occurs, nor has Nanog's function been characterized for skeletal development in vivo. Nanog has also been shown to promote proliferation (Darr et al 2006), and negatively regulates BMP signaling (Suzuki et al 2006) as well (both processes are well known to be involved in skeletal development). Therefore Nanog is a compelling candidate factor to test for its function in regulating Sox9 and skeletal development.
Therefore, I would suggest that an experiment be performed to test to see if Nanog actually binds to its predicted binding sites on the Sox9 locus. I propose that ChIP-seq should be performed using antibodies against Nanog (with IgG antibodies as a negative control) using the limb-explant experimental system described above, using limb buds from embryos at E12.5 (since mesenchymal condensations thru chondrogenesis, stages where Sox9 is known to act, are known to be occuring at this stage). Ideally, Nanog occupancy should be enriched at the loci predicted, but Nanog doesn't necessarily have to bind to the predicted motifs, as long as it binds somewhere within the locus.
Reporter constructs with these sequences where Nanog binds to on the Sox9 locus should be created to see if these sequences are sufficient to confer enhancer activity, and should correlate to where Sox9 is known to be expressed.
Therefore, I would suggest that an experiment be performed to test to see if Nanog actually binds to its predicted binding sites on the Sox9 locus. I propose that ChIP-seq should be performed using antibodies against Nanog (with IgG antibodies as a negative control) using the limb-explant experimental system described above, using limb buds from embryos at E12.5 (since mesenchymal condensations thru chondrogenesis, stages where Sox9 is known to act, are known to be occuring at this stage). Ideally, Nanog occupancy should be enriched at the loci predicted, but Nanog doesn't necessarily have to bind to the predicted motifs, as long as it binds somewhere within the locus.
Reporter constructs with these sequences where Nanog binds to on the Sox9 locus should be created to see if these sequences are sufficient to confer enhancer activity, and should correlate to where Sox9 is known to be expressed.
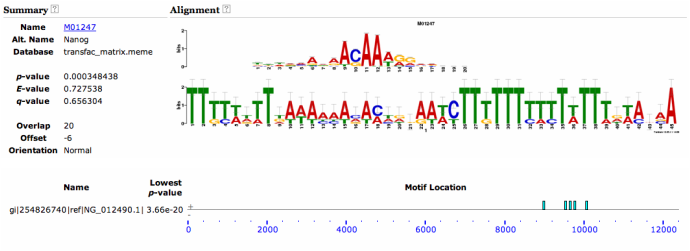
Human Nanog binding motif to Sox9 locus
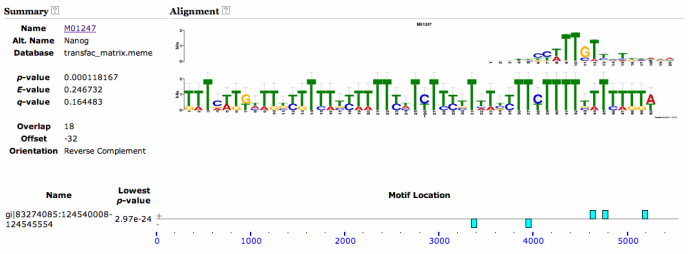
Mouse Nanog binding motif to Sox9 locus
I then looked into seeing if Sox9 and Nanog share any common interactors in their respective networks. I found on STRING that they both share a common interactor in the gene Ctnnb1, which is more well known as Beta-Catenin, which is the transcription factor in canonical Wnt signaling.
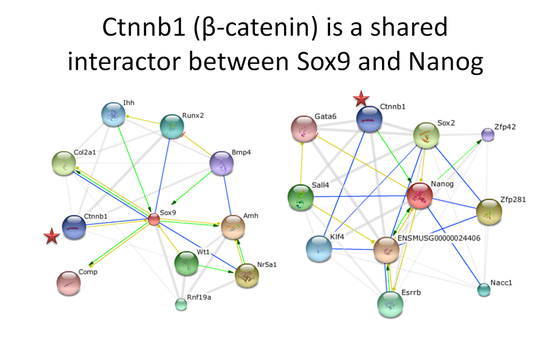
Sox9 has been shown to promote phosphorylation of Beta-Catenin in the nucleus, which is inhibitory on Wnt signaling (Topol et al 2009). Canonical Wnt signaling has been shown to be important in promoting chondrocyte differentiation (Im and Guan 2010) thru Sox9 (Yano et al 2005), and has been shown to promote chondrocyte differentiation and inhibit osteocyte differentiation in skeletal development (Day et al 2005; Hill et al 2005). This suggests some sort of feedback loop where continual Wnt signaling may facilitate further Sox9 expression. Potentially, Nanog may be this intermediary, since Beta-Catenin is able to upregulate Nanog expression when associated with Oct3/4 in embryonic stem cells (Takao et al 2007), and Nanog is able to stimulate Sox9 expression in mesenchymal stem cells (Zheng et al 2010).
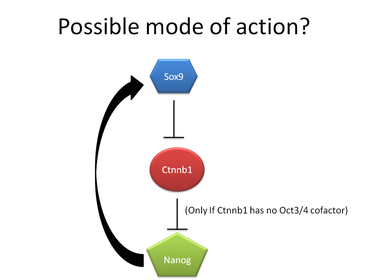
Interestingly enough, in order for Sox9 and Nanog to interact with each other through Beta-Catenin, Beta-Catenin should have inhibitory properties on Nanog, since Sox9 inhibits Beta-Catenin, thus allowing Nanog expression to maintain Sox9 expression (if Beta-Catenin is stimulatory of Nanog, Sox9 would cause a downregulation of Nanog, which then wouldn't be able to maintain Sox9 expression). It should be noted that this interaction of Beta-Catenin being stimulatory required Oct3/4 as a cofactor (Takao et al 2007). Therefore, I hypothesize that without Oct3/4, Beta-Catenin probably has an inhibitory role on Nanog (See generalized model to the right). However, this remains to be tested.
With this information in hand, my second set of experiments I propose would need to determine whether or not Beta-Catenin, without Oct3/4, would be inhibitory of Nanog expression.
To test this, one should transfect Oct3/4 into mesenchymal stem cells that are deficient of Oct3/4 (possibly conditionally knocked-out by cre-recombination) in vitro, with an empty vector as a control. Upon transfection, Nanog levels should be assayed and should decrease in abundance compared to controls. Next, in this population of Oct3/4 transfected cells, siRNA-mediated knockdown of Oct3/4 should be performed. This knockdown of Oct3/4 should result in the restoration of Nanog expression in these MSCs. Furthermore, Sox9 expression should be assayed as well in these samples. I would expect Sox9 expression to be decreased upon Oct3/4 transfection, but should be restored, or even enhanced upon Oct3/4 knockdown.
With this information in hand, my second set of experiments I propose would need to determine whether or not Beta-Catenin, without Oct3/4, would be inhibitory of Nanog expression.
To test this, one should transfect Oct3/4 into mesenchymal stem cells that are deficient of Oct3/4 (possibly conditionally knocked-out by cre-recombination) in vitro, with an empty vector as a control. Upon transfection, Nanog levels should be assayed and should decrease in abundance compared to controls. Next, in this population of Oct3/4 transfected cells, siRNA-mediated knockdown of Oct3/4 should be performed. This knockdown of Oct3/4 should result in the restoration of Nanog expression in these MSCs. Furthermore, Sox9 expression should be assayed as well in these samples. I would expect Sox9 expression to be decreased upon Oct3/4 transfection, but should be restored, or even enhanced upon Oct3/4 knockdown.
Next, I would like to see if Sox9 overexpression would be sufficient to stimulate or inhibit Nanog expression to further test my model. To do this, I will be using Indolactam V to upregulate Sox9 expression via stimulating PKC signaling in embryonic stem cells in vitro as described previously (Chen et al 2009), since there is no evidence of Indolactam V working in vivo. Upon incubation of these ES cells with Indolactam V, levels of protein abundance changes via western blots of Sox9 (as control), Beta-Catenin, and Nanog should be observed, compared to sham-treated ES cells as a control. If the model is to be supported, I would expect that as Sox9 levels increase, Nanog should increase, with Beta-Catenin levels decreasing.
Finally, I would want to test to see if Nanog is even expressed in the relevant embryonic tissues in vivo. This is due to the fact that Nanog has been primarily been characterized in vitro (i.e. various stem cells), with only having epiblast being the in vivo model found so far (Hart et al 2004). Since Nanog and Sox9 seem to be potential interactors, it would be important to see if Sox9 and Nanog appear in the same tissue, i.e. the developing skeleton (using the limb bud as a model).
In order to determine whether or not Nanog is found to be expressed in the developing skeleton (E10.5 and/or E12.5), I would like to generate a transgenic mouse for a GFP reporter fused to the promoter of Nanog, which should mark with GFP where Nanog is expressed in the embryo. I would expect Nanog-GFP expression to appear where the mesenchymal condensations are to be occuring. Upon whole-mount in situ hybridizations and immunohistochemistry of Beta-Catenin and Sox9 in these limb buds, these should colocalize with Nanog-GFP at predicted levels of expression in the limb bud. Thus, this would indicate whether or not Nanog would even be physiologically relevant in vivo, or if it is just an artifact of the culturing conditions.
References
Johnson DS, Mortazavi A, Myers RM, Wold B. (2007) Genome-wide mapping of in vivo protein–DNA interactions. Science. 316: 1497–1502
Jothi R, Cuddapah S, Barski A, Cui K, Zhao K. (2008) Genome-wide identification of in vivo protein–DNA binding sites from ChIP-Seq data. Nucleic Acids Res. 36(16) 5221–5231
Oh CD, Maity SN, Lu JF, Zhang J, Liang S, Coustry F, de Crombrugghe B, Yasuda H. (2010). Identification of SOX9 Interaction Sites in the Genome of Chondrocytes. PLoS One. 5(4): e10113
Mitsui K, Tokuzawa Y, Itoh H, Segawa K, Murakami M, Takahashi K, Maruyama M, Maeda M, Yamanaka S. The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell. 2003 May 30;113(5):631-42.
Zheng H, Gourronc F, Buckwalter JA, Martin JA. Nanog maintains human chondrocyte phenotype and function in vitro. J Orthop Res. 2010 Apr;28(4):516-21
Darr H, Mayshar Y, Benvenisty N. Overexpression of NANOG in human ES cells enables feeder-free growth while inducing primitive ectoderm features. Development. 2006 Mar;133(6):1193-201
Suzuki A, Raya A, Kawakami Y, Morita M, Matsui T, Nakashima K, Gage FH, Rodríguez-Esteban C, Izpisúa Belmonte JC. Nanog binds to Smad1 and blocks bone morphogenetic protein-induced differentiation of embryonic stem cells. Proc Natl Acad Sci U S A. 2006. 103(27):10294-9.
Topol L, Chen W, Song H, Day TF, Yang Y. Sox9 inhibits Wnt signaling by promoting beta-catenin phosphorylation in the nucleus. J Biol Chem. 2009 Jan 30;284(5):3323-33.
Yano F, Kugimiya F, Ohba S, Ikeda T, Chikuda H, Ogasawara T, Ogata N, Takato T, Nakamura K, Kawaguchi H, Chung UI. The canonical Wnt signaling pathway promotes chondrocyte differentiation in a Sox9-dependent manner. Biochem Biophys Res Commun. 2005 Aug 12;333(4):1300-8.
Takao Y, Yokota T, Koide H. Beta-catenin up-regulates Nanog expression through interaction with Oct-3/4 in embryonic stem cells.Biochem Biophys Res Commun. 2007 Feb 16;353(3):699-705.
Day TF, Guo X, Garrett-Beal L, Yang Y. Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell. 2005 May;8(5):739-50.
Hill TP, Später D, Taketo MM, Birchmeier W, Hartmann C. Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev Cell. 2005 May;8(5):727-38.
Im GI, Quan Z. The effects of Wnt inhibitors on the chondrogenesis of human mesenchymal stem cells. Tissue Eng Part A. 2010 Jul;16(7):2405-13.
Chen, S., Borowiak, M., Fox, J.L., Maehr, R., Osafune, K., Davidow, L., Lam, K., Peng, L.F., Schreiber, S.L., Rubin, L.L., Melton, D. (2009) A small molecule that directs differentiation of human ESCs into the pancreatic lineage. Nat Chem Biol 5: 258-65.
Hart AH, Hartley L, Ibrahim M, Robb L. Identification, cloning and expression analysis of the pluripotency promoting Nanog genes in mouse and human. Dev Dyn. 2004 May;230(1):187-98.
Jothi R, Cuddapah S, Barski A, Cui K, Zhao K. (2008) Genome-wide identification of in vivo protein–DNA binding sites from ChIP-Seq data. Nucleic Acids Res. 36(16) 5221–5231
Oh CD, Maity SN, Lu JF, Zhang J, Liang S, Coustry F, de Crombrugghe B, Yasuda H. (2010). Identification of SOX9 Interaction Sites in the Genome of Chondrocytes. PLoS One. 5(4): e10113
Mitsui K, Tokuzawa Y, Itoh H, Segawa K, Murakami M, Takahashi K, Maruyama M, Maeda M, Yamanaka S. The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell. 2003 May 30;113(5):631-42.
Zheng H, Gourronc F, Buckwalter JA, Martin JA. Nanog maintains human chondrocyte phenotype and function in vitro. J Orthop Res. 2010 Apr;28(4):516-21
Darr H, Mayshar Y, Benvenisty N. Overexpression of NANOG in human ES cells enables feeder-free growth while inducing primitive ectoderm features. Development. 2006 Mar;133(6):1193-201
Suzuki A, Raya A, Kawakami Y, Morita M, Matsui T, Nakashima K, Gage FH, Rodríguez-Esteban C, Izpisúa Belmonte JC. Nanog binds to Smad1 and blocks bone morphogenetic protein-induced differentiation of embryonic stem cells. Proc Natl Acad Sci U S A. 2006. 103(27):10294-9.
Topol L, Chen W, Song H, Day TF, Yang Y. Sox9 inhibits Wnt signaling by promoting beta-catenin phosphorylation in the nucleus. J Biol Chem. 2009 Jan 30;284(5):3323-33.
Yano F, Kugimiya F, Ohba S, Ikeda T, Chikuda H, Ogasawara T, Ogata N, Takato T, Nakamura K, Kawaguchi H, Chung UI. The canonical Wnt signaling pathway promotes chondrocyte differentiation in a Sox9-dependent manner. Biochem Biophys Res Commun. 2005 Aug 12;333(4):1300-8.
Takao Y, Yokota T, Koide H. Beta-catenin up-regulates Nanog expression through interaction with Oct-3/4 in embryonic stem cells.Biochem Biophys Res Commun. 2007 Feb 16;353(3):699-705.
Day TF, Guo X, Garrett-Beal L, Yang Y. Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell. 2005 May;8(5):739-50.
Hill TP, Später D, Taketo MM, Birchmeier W, Hartmann C. Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev Cell. 2005 May;8(5):727-38.
Im GI, Quan Z. The effects of Wnt inhibitors on the chondrogenesis of human mesenchymal stem cells. Tissue Eng Part A. 2010 Jul;16(7):2405-13.
Chen, S., Borowiak, M., Fox, J.L., Maehr, R., Osafune, K., Davidow, L., Lam, K., Peng, L.F., Schreiber, S.L., Rubin, L.L., Melton, D. (2009) A small molecule that directs differentiation of human ESCs into the pancreatic lineage. Nat Chem Biol 5: 258-65.
Hart AH, Hartley L, Ibrahim M, Robb L. Identification, cloning and expression analysis of the pluripotency promoting Nanog genes in mouse and human. Dev Dyn. 2004 May;230(1):187-98.
